
手機掃碼訪問本站

微信咨詢
依澤替米貝(Ezetimibe)是由美國先靈葆雅制藥公司(Schering-Plough和默克公司(Merck)聯合研發的一款新型降脂藥。藥理作用研究表明:依澤替米貝是一種新型的選擇性膽固醇吸收抑制劑,可以通過影響小腸刷狀緣攝取和轉運膽固醇微膠粒的載體活性,從而抑制腸壁吸收食物和膽汁中的膽固醇和植物固醇而降低體內膽固醇的水平。實驗證實:依澤替米貝對小腸和肝臟膽固醇的合成沒有直接影響,只能通過阻止腸壁囊泡中的外源性膽固醇轉運至細胞內膽固醇池,抑制外援性膽固醇轉移到淋巴,但并不影響新合成的膽固醇(內源性)合成到小腸的脂蛋白中。
臨床上,依澤替米貝被用于高膽固醇血癥的治療,可以顯著降低高血脂癥患者的血漿低密度脂蛋白膽固醇(LDL-C)、總膽固醇(TC)和齲齒類對甘油三酯(TG)水平,并升高高密度脂蛋白膽固醇(HDL-C)水平。當與其他汀類藥物聯合使用時,可同時抑制膽固醇的內源和外源代謝途徑,可以更好地控制高血脂癥患者的血脂水平,取得顯著地降低血脂和抗動脈粥樣硬化效果。依澤替米貝同時也可用于兩種不常見的高血脂癥治療:純合子家族遺傳性高膽固醇血癥和純合子谷固醇血癥。
依澤替米貝于2002年10月經過美國FDA批準上市,商品名“益適純(Zetia/Ezetrol)。目前已經在超過90個國家和地區上市銷售。依澤替米貝在2006年經過國家食品藥品監督管理局(SFDA)批準在中國獲得注冊銷售,2007年下半年開始正式起步銷售,目前已在多地納入了醫保目錄中。
目前已經報道的用于依澤替米貝合成的方法有多種,下面我們對幾種合成方法進行介紹:
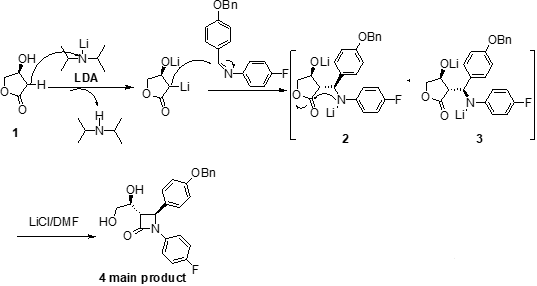
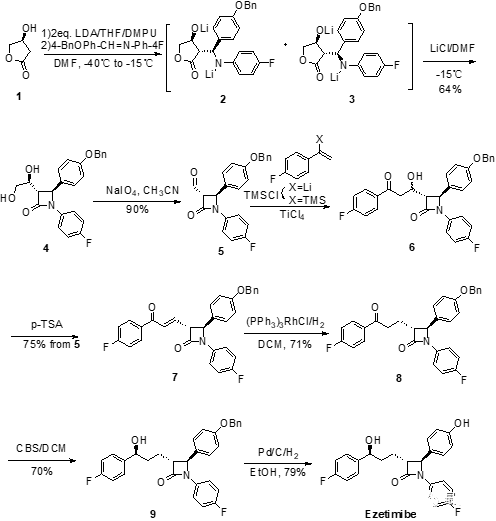
首先,依澤替米貝可以通過利用市售的(S)-3-羥基-γ-內酯1一步合成非對映選擇性的反式-β-內酰胺4開始。原料1在四氫呋喃中用二異丁基胺基鋰(LDA)處理后加入N-(4-芐氧基)甲苯-N-(4-氟)苯基亞胺和N,N-二甲基丙烯脲(DMPU)可得到2和3的混合物,其中2為主要產物,其比例為2:3=79:21。DMPA是一種優良的有機溶劑,毒性極低,耐光熱,耐酸堿,可溶解各種有機物,無機物,也可作為高反應性親核試劑和堿的助溶劑,使用過程中經常可代替具有致癌性的六甲基磷酰三胺(HMPA)。2和3由于形成了穩定的二價負離子鋰鹽聚合物而無法直接閉環得到中間體4。而當加入了N,N-二甲基甲酰胺(DMF)的氯化鋰(LiCl)溶液后,鋰鹽聚合物發生溶解,中間體分子內發生閉環得到內酰胺主產物4(trans:cis=95:5)。可能由于中間體3胺基和酯羰基空間上距離比較遠,顯負電性的氮難以與羰基發生親核反應,所以其對應的四元環內酰胺產物更加難以生成。
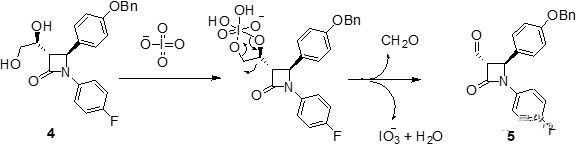
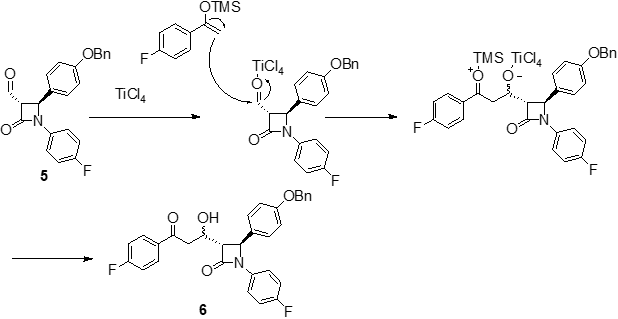
內酰胺中間體4結構中的鄰二醇結構經過高碘酸鈉氧化后發生碳-碳鍵斷裂得到醛中間體5。利用相山-阿德爾縮合反應(Mukaiyama-Aldol condensation),在Lewis酸TiCl4活化作用下,4-氟苯乙烯醇的四甲基硅烷化衍生物和中間體5的醛基發生親核加成反應得到縮合產物6。
傳統的Aldol縮合反應也稱為醇醛縮合反應,是一個烯醇離子和羰基化合物縮合而形成一個β-羥基化合物的反應,有時可接著脫水生成共軛的烯酮產物。通常的醇醛縮合反應往往存在選擇性差,副產物多的缺點,大大限制了其應用范圍。1973年,日本化學家相山光昭(Teruaki Mukaiyama)對Aldol縮合反應進行了改進,將烯醇負離子轉化為硅醚結構進行穩定和分離,然后再用其與醛酮進行反應制備β-羥基化合物或共軛烯酮。經過硅醚化的烯醇化合物其親核反應性能減弱,需要在Lewis酸或堿的幫助下才能實現與醛酮的反應。不經過手性控制的條件下,相山-阿德爾醇醛縮合反應一般得到的是外消旋混合物。當然,目前也有很多文獻和工作報道了對映選擇性控制的不對稱相山-阿德爾反應。
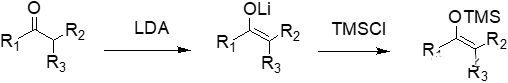
得到的外消旋混合物6不經過分離處理,直接加對甲苯磺酸(p-TSA),β-羥基脫水生成了α,β-不飽和酮7。中間體7的碳-碳雙鍵用威爾金森催化劑((PPh3)3RhCl)和氫氣進行催化氫化還原,得到中間體8。中間體8結構中的酮羰基再經過CBS對映選擇性催化還原后得到帶醇羥基的中間體9。
CBS還原反應即為科里-巴克什-柴田還原反應(Corey-Bakshi-Shibata(CBS)Reduction)。該反應所用的CBS催化劑是不對稱還原反應中重要的手性催化劑,是利用脯氨酸為原料合成得到的手性硼雜惡唑烷硼烷催化劑,在硼烷存在下,可對酮進行不對稱催化還原。不管是環狀的酮還是鏈狀酮,都可以被還原成高對應選擇性的醇,其產物的手性構型和催化劑的構型有關,R-構型的CBS催化劑往往得到高選擇性的S-構型的產物。研究還發現,CBS還原反應受到溫度的影響,通過控制溫度可以有效的控制反應進行和產物的對應選擇性。
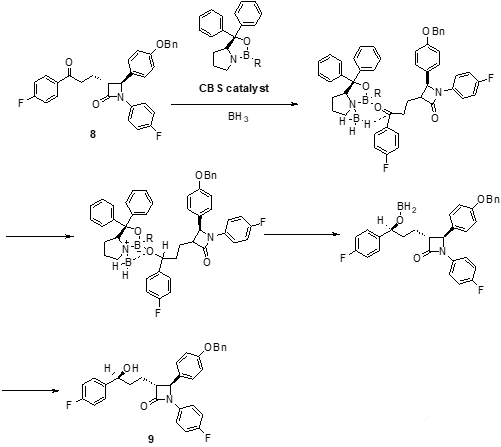
中間體9再經過鈀碳(Pd/C)催化氫化之后脫去芐基(Bn)并最終得到依澤替米貝。
除了上述方法,中間體7也可以用Pd/C和氫氣(H2)直接一鍋法進行雙鍵的加氫還原和芐基的脫除得到中間體10,中間體10再經過六甲基二硅脲(BSU)的處理,將結構中的酚羥基進行硅烷化保護,再進一步用CBS對映選擇性還原結構中的酮得到醇羥基中間體12,12經過HCl處理后脫去三甲基硅基得到依澤替米貝。中間體10到依澤替米貝的三步合成可以通過一鍋法高效實現。

除了上述兩種合成方法之外,第三種合成方法路線如下:
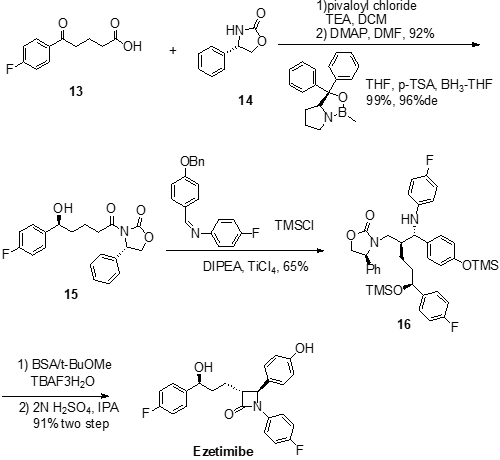
原料13首先和三甲基乙酰氯(pivaloyl chloride)反應,將羧基酰氯化,三乙胺(TEA)在起活化促進反應的作用。生成酰氯活性中間體后,進一步和手性輔助結構14中的胺基反應生成酮基酰胺中間體,反應中用4-二甲氨基吡啶(DMAP)催化促進反應進行。生成的產物進一步用CBS進行對映選擇性催化還原,將酮羰基還原為醇羥基,得到中間體15,該反應過程選擇了對甲苯磺酸(p-TSA)作為Lewis酸。以N,N-二異丙基乙胺(DIPEA)做堿,中間體15在四氯化鈦(TiCl4)催化下與N-(4-芐氧基)甲苯-N-(4-氟)苯基亞胺發生親電加成反應(類似于Michael加成反應),生成的中間體進一步用三甲基氯硅烷(TMSCl)處理,將活性羥基進行硅烷化保護得到中間體16。中間體16首先用牛血清蛋白(BSA)和四丁基氟化銨(TBAF)處理,水解后發生分子內成環,生成四元環內酰胺中間體,之后再用稀硫酸溶液處理,脫去分子結構中的三甲基硅基保護基(TMS)并最終以91%的兩步總收率得到目標產物依澤替米貝。
Jin Li and Kevin K.-C. Liu Synthetic Approaches to the 2002 New Drugs.Mini-Reviewsin Medicinal Chemistry, 2004, 4, 207-233.